IgG4-related sclerosing cholangitis (IgG4-SC) is known in the adult patients as a steroid-responsive biliary disease, frequently associated with autoimmune pancreatitis; The diagnosis of IgG4-SC may be difficult to differentiate from primary sclerosing cholangitis (PSC) or cholangiocarcinoma; This entity is been described in the absence of pancreatic implication. It is defined by high level of serum IgG4 in contrast to primary sclerosing cholangitis. It’is morphologically characterized by dense lymphoplasmacellular infiltration, particularly IgG4+ plasma cells and CD4+ T cells and extensive fibrosis in bile duct. In patients with IgG4-related sclerosing cholangitis, response to steroid therapy is high; in patients with PSC corticosteroid therapy is unsuccessful. An Early recognition of IgG4-SC can save patients from potential harmful and unnecessary surgical interventions. In the literature, cholangiocarcinoma in patients with IgG4- related sclerosing cholangitis was not described, whereas cholangiocarcinoma develops in up to 10-30% of patients with PSC. We present the case of a 3 years old child with features of sclerosing IgG4 cholangitis with asymptomatic elevation in liver enzymes, bile duct strictures on imaging, characteristic pathology findings, elevated serum IgG4, without signs of pancreatic involvement, and excellent response to corticosteroids. Pediatric gastroenterologists and hepatologists, as well as pediatric hepatopathologists, need to be aware of IgG4-SC as a disease entity.
| Published in | American Journal of Pediatrics (Volume 10, Issue 3) |
| DOI | 10.11648/j.ajp.20241003.19 |
| Page(s) | 158-162 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2024. Published by Science Publishing Group |
IgG4 Level, Cholangitis, Steroid, Lymphoplasmacytic Infiltration, Child
AST (U/l) | ALT (U/l) | TB (mg/l) | BC (mg/l) | LIPASE (U/l) | IgG4 (U/l) | Prednisone | Azathioprine | Ursodeoxycholic acid | |
|---|---|---|---|---|---|---|---|---|---|
At initial presentation | 458 | 362 | 80 | 60 | 17 | 2.7 | 1m/kg/d | - | 15mg/kg/d |
6 weeks after diagnosis | 382 | 295 | 73.7 | 50.5 | 12 | - | 1m/kg/d | 1.5mg/kg/d | 15mg/kg/d |
10 weeks after diagnosis | 292 | 235 | 48 | 29 | 20 | 2.6 | 1m/kg/d | 1.5mg/kg/d | 15mg/kg/d |
6 months after diagnosis | 242 | 136 | 42 | 23 | 23 | 2.6 | 0.5m/kg/d | 1.5mg/kg/d | 15mg/kg/d |
8 months after diagnosis | 150 | 120 | 41 | 21 | 16 | 2, 5 | 0.1mg/kg/d | 1.5mg/kg/d | 15mg/kg/d |
SC | Sclerosing Cholangitis |
PSC | Primary Sclerosing Cholangitis |
| [1] | Einar Bjornson. immunoglobulin g4-associated cholangitis. current opinion in. 2008, 24: 389-394. |
| [2] | Faiz Karim, Jan Loeffen, Wichor Bramer: Lauren Westenberg, Rob Verdijk, Martin van Hagenand Jan van Laar. IgG4-related disease: a systematic review of this unrecognized disease in pediatrics. Pediatric. Rheumatology 14. 18: |
| [3] | Chien-Ting Hsu Yung-Ming Jeng, Jia-Feng Wu: Immunoglobulin G4-related sclerosing cholangitis in a 3 years of age boy. Adv Dig Med. 2021. 8: 59-63. |
| [4] | Olivier CHAZOUILLERES: Primary sclerosing cholangitis: diagnosis and follow-up. POST’U. 2020, |
| [5] | Hakima Abid, Moulaye El Hacen Horma Babana El Alaoui, Moulay Youssef Alaoui Lamrani, et al.: AndNourdin Aqodad. IgG4 disease apropos of 3 cases. an Afr Med J. 2020, 28: 364. |
| [6] | Kamonchanok Phaopraphat, Pintip Ngamjanyaporn, Pongthorn Narongroeknawin, Nuntana Kasitanon, Wanruchada Katchamart: Clinical manifestations, clinical course, and outcomes of immunoglobulin G4-related disease. Int J Rheum Dis. 2020, 23: 1468-1473. |
| [7] | Amaar ghazale, suresh t. Chari, lizhi zhang, et al.: Pearson, bret t. Petersen, santhi swaroop vege, keith lindor and michael b. Farnel. Immunoglobulin. 4, 2008: 706-715. |
| [8] | Thumper Zhang, Yiyi Gong, Zheng Liu, et al.: Efficacy and safety of iguratimod plus corticosteroid as bridge therapy in treating mild IgG4-related diseases: A prospective clinical trial. Int J Rheum Dis2019 Aug. 22: 1479-1488. |
| [9] | Tanaka A., Tazuma S., Okazaki K., Tsubouchi H., Inui K., Takikawa H. Nationwide survey for primary sclerosing cholangitis and igg4-related sclerosing cholangitis in Japan. Journal of Hepato-Biliary-Pancreatic Sciences. 2014; 21(1): 43–50. |
| [10] | Manganis C. D., Chapman R. W., Culver E. L. Review of primary sclerosing cholangitis with increased igg4 levels. World Journal of Gastroenterology. 2020; 26(23): 3126–3144. |
| [11] | Zen Y., Kawakami H., Kim J. H. Igg4-related sclerosing cholangitis: all we need to know. Journal of Gastroenterology. 2016; 51(4): 295–312. |
| [12] | Nakazawa T., Kamisawa T., Okazaki K., et al. Clinical diagnostic criteria for IgG4-related sclerosing cholangitis 2020. Journal of Hepato-Biliary-Pancreatic Sciences. 2021; 28(3): 235–242. |
| [13] | Moon S.-H., Kim M.-H., Lee J. K., et al. Development of a scoring system for differentiating igg4-related sclerosing cholangitis from primary sclerosing cholangitis. Journal of Gastroenterology. 2017; 52(4): 483–493. |
APA Style
Sabbahia, D. B., Msaaf, H., Atrassi, M., Moukhlis, S., Bennani, N., et al. (2024). IgG4 Sclerosing Cholangitis: Entity Rarely Described in Children. American Journal of Pediatrics, 10(3), 158-162. https://doi.org/10.11648/j.ajp.20241003.19
ACS Style
Sabbahia, D. B.; Msaaf, H.; Atrassi, M.; Moukhlis, S.; Bennani, N., et al. IgG4 Sclerosing Cholangitis: Entity Rarely Described in Children. Am. J. Pediatr. 2024, 10(3), 158-162. doi: 10.11648/j.ajp.20241003.19
@article{10.11648/j.ajp.20241003.19,
author = {Dalal Ben Sabbahia and Halima Msaaf and Meriem Atrassi and Sara Moukhlis and Nissrine Bennani and Abdelhak Abkari},
title = {IgG4 Sclerosing Cholangitis: Entity Rarely Described in Children
},
journal = {American Journal of Pediatrics},
volume = {10},
number = {3},
pages = {158-162},
doi = {10.11648/j.ajp.20241003.19},
url = {https://doi.org/10.11648/j.ajp.20241003.19},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajp.20241003.19},
abstract = {IgG4-related sclerosing cholangitis (IgG4-SC) is known in the adult patients as a steroid-responsive biliary disease, frequently associated with autoimmune pancreatitis; The diagnosis of IgG4-SC may be difficult to differentiate from primary sclerosing cholangitis (PSC) or cholangiocarcinoma; This entity is been described in the absence of pancreatic implication. It is defined by high level of serum IgG4 in contrast to primary sclerosing cholangitis. It’is morphologically characterized by dense lymphoplasmacellular infiltration, particularly IgG4+ plasma cells and CD4+ T cells and extensive fibrosis in bile duct. In patients with IgG4-related sclerosing cholangitis, response to steroid therapy is high; in patients with PSC corticosteroid therapy is unsuccessful. An Early recognition of IgG4-SC can save patients from potential harmful and unnecessary surgical interventions. In the literature, cholangiocarcinoma in patients with IgG4- related sclerosing cholangitis was not described, whereas cholangiocarcinoma develops in up to 10-30% of patients with PSC. We present the case of a 3 years old child with features of sclerosing IgG4 cholangitis with asymptomatic elevation in liver enzymes, bile duct strictures on imaging, characteristic pathology findings, elevated serum IgG4, without signs of pancreatic involvement, and excellent response to corticosteroids. Pediatric gastroenterologists and hepatologists, as well as pediatric hepatopathologists, need to be aware of IgG4-SC as a disease entity.
},
year = {2024}
}
TY - JOUR T1 - IgG4 Sclerosing Cholangitis: Entity Rarely Described in Children AU - Dalal Ben Sabbahia AU - Halima Msaaf AU - Meriem Atrassi AU - Sara Moukhlis AU - Nissrine Bennani AU - Abdelhak Abkari Y1 - 2024/08/30 PY - 2024 N1 - https://doi.org/10.11648/j.ajp.20241003.19 DO - 10.11648/j.ajp.20241003.19 T2 - American Journal of Pediatrics JF - American Journal of Pediatrics JO - American Journal of Pediatrics SP - 158 EP - 162 PB - Science Publishing Group SN - 2472-0909 UR - https://doi.org/10.11648/j.ajp.20241003.19 AB - IgG4-related sclerosing cholangitis (IgG4-SC) is known in the adult patients as a steroid-responsive biliary disease, frequently associated with autoimmune pancreatitis; The diagnosis of IgG4-SC may be difficult to differentiate from primary sclerosing cholangitis (PSC) or cholangiocarcinoma; This entity is been described in the absence of pancreatic implication. It is defined by high level of serum IgG4 in contrast to primary sclerosing cholangitis. It’is morphologically characterized by dense lymphoplasmacellular infiltration, particularly IgG4+ plasma cells and CD4+ T cells and extensive fibrosis in bile duct. In patients with IgG4-related sclerosing cholangitis, response to steroid therapy is high; in patients with PSC corticosteroid therapy is unsuccessful. An Early recognition of IgG4-SC can save patients from potential harmful and unnecessary surgical interventions. In the literature, cholangiocarcinoma in patients with IgG4- related sclerosing cholangitis was not described, whereas cholangiocarcinoma develops in up to 10-30% of patients with PSC. We present the case of a 3 years old child with features of sclerosing IgG4 cholangitis with asymptomatic elevation in liver enzymes, bile duct strictures on imaging, characteristic pathology findings, elevated serum IgG4, without signs of pancreatic involvement, and excellent response to corticosteroids. Pediatric gastroenterologists and hepatologists, as well as pediatric hepatopathologists, need to be aware of IgG4-SC as a disease entity. VL - 10 IS - 3 ER -
The Department of Pediatrics III, Unit of Pediatric Gastroenterology and Hepatology, Children’s Hospital A. Harouchi, Casablanca, Morocco
The Department of Pediatrics III, Unit of Pediatric Gastroenterology and Hepatology, Children’s Hospital A. Harouchi, Casablanca, Morocco
The Department of Pediatrics III, Unit of Pediatric Gastroenterology and Hepatology, Children’s Hospital A. Harouchi, Casablanca, Morocco
Central Service of Pathological Anatomy, Ibn Rochd’s Hospital, Casablanca, Morocco
Central Service of Pathological Anatomy, Ibn Rochd’s Hospital, Casablanca, Morocco
The Department of Pediatrics III, Unit of Pediatric Gastroenterology and Hepatology, Children’s Hospital A. Harouchi, Casablanca, Morocco
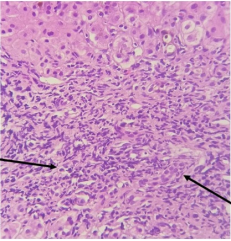
Figure 1. Histological image stained with hematein eosin (Magnification x40) which shows the portal inflammatory infiltrate mainly lymphoplasmacytic associated with destruction of bile ducts.
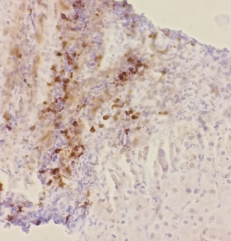
Figure 2. Immunohistochemical study with CD138 Magnification x 40) highlighting the presence of plasma cells taking on a membrane stain.
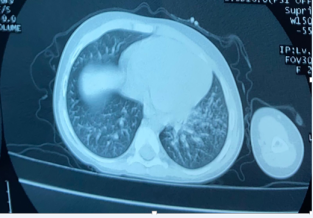
Figure 3. Micronodular infiltrate with centrilobular left basal bronchiolar distribution.
Information

