Background: Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) and Hypertrophic Cardiomyopathy (HCM) are distinct inherited cardiac disorders that represent leading causes of sudden cardiac death, particularly in young adults. While both conditions display autosomal dominant inheritance patterns, they typically involve different genetic mutations and pathophysiological mechanisms. Case report: We present a rare case of a 45-year-old male with ventricular tachycardia who demonstrated concurrent phenotypic features of both ARVC and HCM on comprehensive cardiac evaluation. Electrocardiography showed epsilon waves characteristic of ARVC, while cardiac magnetic resonance imaging (CMR) revealed right ventricular dilatation consistent with ARVC alongside mid-ventricular hypertrophic obstructive cardiomyopathy (HOCM). This unusual phenotypic overlap highlights the importance of comprehensive multimodality cardiac imaging and raises intriguing questions about potential genetic and molecular intersections between these cardiomyopathies. Conclusion: This case highlights the importance of comprehensive multimodality cardiac imaging in identifying complex structural abnormalities and raises questions about potential genetic and molecular intersections between ARVC and HCM. Our case also adds to the extremely limited literature documenting the co-occurrence of these conditions and underscores the value of CMR in identifying complex structural cardiac abnormalities. Management of patients with this rare phenotypic overlap presents unique challenges, requiring careful consideration of risk stratification and medical therapy. Further research may provide valuable insights into the pathophysiology and optimal management strategies for patients with overlapping cardiomyopathic phenotypes.
| Published in | Cardiology and Cardiovascular Research (Volume 9, Issue 2) |
| DOI | 10.11648/j.ccr.20250902.14 |
| Page(s) | 64-68 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Arrhythmogenic Right Ventricular Cardiomyopathy, Hypertrophic Cardiomyopathy, Mid-ventricular Obstruction, Ventricular Tachycardia, Epsilon Wave, Cardiac Magnetic Resonance Imaging
ARVC | Arrhythmogenic Right Ventricular Cardiomyopathy |
HCM | Hypertrophic Cardiomyopathy |
SCD | Sudden Cardiac Death |
LGE | Late Gadolinium Enhancement |
CMR | Cardiac Magnetic Resonance |
HOCM | Hypertrophic Obstructive Cardiomyopathy |
ECV | Extracellular Volume |
| [1] | Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017; 121(7): 784-802. |
| [2] | Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J Am Coll Cardiol. 2014; 64(1): 83-99. |
| [3] | Finocchiaro G, Papadakis M, Robertus JL, et al. Etiology of Sudden Death in Sports: Insights From a United Kingdom Regional Registry. J Am Coll Cardiol. 2016; 67(18): 2108-2115. |
| [4] | Groeneweg JA, Bhonsale A, James CA, et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet. 2015; 8(3): 437-446. |
| [5] | Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015; 65(12): 1249-1254. |
| [6] | Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008; 52(25): 2175-2187. |
| [7] | Rampazzo A, Calore M, van Hengel J, van Roy F. Intercalated discs and arrhythmogenic cardiomyopathy. Circ Cardiovasc Genet. 2014; 7(6): 930-940. |
| [8] |
Mashego B, Nethononda MR. Unusual Combination Hypertrophic Cardiomyopathy and Arrhythmogenic Cardiomyopathy Phenotype. Austin J Radiol. 2020; 7(2): 1110. URL:
https://austinpublishinggroup.com/radiology/austin-j-radiol-7-2.php |
| [9] | Towbin JA. Inherited cardiomyopathies. Circ J. 2014; 78(10): 2347-2356. |
| [10] | Gerull B. Desmosomal Cadherin-2 and Arrhythmogenic Cardiomyopathy. Card Electrophysiol Clin. 2020; 12(2): 201-208. |
| [11] | Ho CY, Day SM, Ashley EA, et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018; 138(14): 1387-1398. |
| [12] | Efthimiadis GK, Pagourelias ED, Hadjimiltiades S, et al. Clinical characteristics and natural history of hypertrophic cardiomyopathy with midventricular obstruction. Circ J. 2013; 77(9): 2366-2374. |
| [13] | Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol. 2013; 61(19): 1945-1948. |
| [14] | Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2004; 13(4): 185-194. |
| [15] | Chelko SP, Asimaki A, Andersen P, et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 2016; 1(5): e85923. |
| [16] | Bauce B, Nava A, Beffagna G, et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010; 7(1): 22-29. |
| [17] | Mazzarotto F, Olivotto I, Boschi B, et al. Contemporary Insights Into the Genetics of Hypertrophic Cardiomyopathy: Toward a New Era in Clinical Testing? J Am Heart Assoc. 2020; 9(8): e015473. |
| [18] | Pinto YM, Elliott PM, Arbustini E, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016; 37(23): 1850-1858. |
APA Style
Kapoor, A., Chopra, A., Singh, H. P. (2025). Concurrent Arrhythmogenic Right Ventricular Cardiomyopathy and Hypertrophic Cardiomyopathy: A Rare Phenotypic Overlap. Cardiology and Cardiovascular Research, 9(2), 64-68. https://doi.org/10.11648/j.ccr.20250902.14
ACS Style
Kapoor, A.; Chopra, A.; Singh, H. P. Concurrent Arrhythmogenic Right Ventricular Cardiomyopathy and Hypertrophic Cardiomyopathy: A Rare Phenotypic Overlap. Cardiol. Cardiovasc. Res. 2025, 9(2), 64-68. doi: 10.11648/j.ccr.20250902.14
@article{10.11648/j.ccr.20250902.14,
author = {Atul Kapoor and Arun Chopra and Harinder Pal Singh},
title = {Concurrent Arrhythmogenic Right Ventricular Cardiomyopathy and Hypertrophic Cardiomyopathy: A Rare Phenotypic Overlap
},
journal = {Cardiology and Cardiovascular Research},
volume = {9},
number = {2},
pages = {64-68},
doi = {10.11648/j.ccr.20250902.14},
url = {https://doi.org/10.11648/j.ccr.20250902.14},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ccr.20250902.14},
abstract = {Background: Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) and Hypertrophic Cardiomyopathy (HCM) are distinct inherited cardiac disorders that represent leading causes of sudden cardiac death, particularly in young adults. While both conditions display autosomal dominant inheritance patterns, they typically involve different genetic mutations and pathophysiological mechanisms. Case report: We present a rare case of a 45-year-old male with ventricular tachycardia who demonstrated concurrent phenotypic features of both ARVC and HCM on comprehensive cardiac evaluation. Electrocardiography showed epsilon waves characteristic of ARVC, while cardiac magnetic resonance imaging (CMR) revealed right ventricular dilatation consistent with ARVC alongside mid-ventricular hypertrophic obstructive cardiomyopathy (HOCM). This unusual phenotypic overlap highlights the importance of comprehensive multimodality cardiac imaging and raises intriguing questions about potential genetic and molecular intersections between these cardiomyopathies. Conclusion: This case highlights the importance of comprehensive multimodality cardiac imaging in identifying complex structural abnormalities and raises questions about potential genetic and molecular intersections between ARVC and HCM. Our case also adds to the extremely limited literature documenting the co-occurrence of these conditions and underscores the value of CMR in identifying complex structural cardiac abnormalities. Management of patients with this rare phenotypic overlap presents unique challenges, requiring careful consideration of risk stratification and medical therapy. Further research may provide valuable insights into the pathophysiology and optimal management strategies for patients with overlapping cardiomyopathic phenotypes.
},
year = {2025}
}
TY - JOUR T1 - Concurrent Arrhythmogenic Right Ventricular Cardiomyopathy and Hypertrophic Cardiomyopathy: A Rare Phenotypic Overlap AU - Atul Kapoor AU - Arun Chopra AU - Harinder Pal Singh Y1 - 2025/06/18 PY - 2025 N1 - https://doi.org/10.11648/j.ccr.20250902.14 DO - 10.11648/j.ccr.20250902.14 T2 - Cardiology and Cardiovascular Research JF - Cardiology and Cardiovascular Research JO - Cardiology and Cardiovascular Research SP - 64 EP - 68 PB - Science Publishing Group SN - 2578-8914 UR - https://doi.org/10.11648/j.ccr.20250902.14 AB - Background: Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) and Hypertrophic Cardiomyopathy (HCM) are distinct inherited cardiac disorders that represent leading causes of sudden cardiac death, particularly in young adults. While both conditions display autosomal dominant inheritance patterns, they typically involve different genetic mutations and pathophysiological mechanisms. Case report: We present a rare case of a 45-year-old male with ventricular tachycardia who demonstrated concurrent phenotypic features of both ARVC and HCM on comprehensive cardiac evaluation. Electrocardiography showed epsilon waves characteristic of ARVC, while cardiac magnetic resonance imaging (CMR) revealed right ventricular dilatation consistent with ARVC alongside mid-ventricular hypertrophic obstructive cardiomyopathy (HOCM). This unusual phenotypic overlap highlights the importance of comprehensive multimodality cardiac imaging and raises intriguing questions about potential genetic and molecular intersections between these cardiomyopathies. Conclusion: This case highlights the importance of comprehensive multimodality cardiac imaging in identifying complex structural abnormalities and raises questions about potential genetic and molecular intersections between ARVC and HCM. Our case also adds to the extremely limited literature documenting the co-occurrence of these conditions and underscores the value of CMR in identifying complex structural cardiac abnormalities. Management of patients with this rare phenotypic overlap presents unique challenges, requiring careful consideration of risk stratification and medical therapy. Further research may provide valuable insights into the pathophysiology and optimal management strategies for patients with overlapping cardiomyopathic phenotypes. VL - 9 IS - 2 ER -
Department of Radiology, Advanced Diagnostics, Department of Cardiology Fortis Hospital, Amritsar, India
Department of Radiology, Advanced Diagnostics, Department of Cardiology Fortis Hospital, Amritsar, India
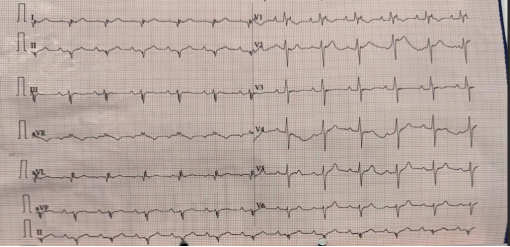
Figure 1. Electrocardiogram showing presence of epsilon waves in I-III and aVL, avF leads.
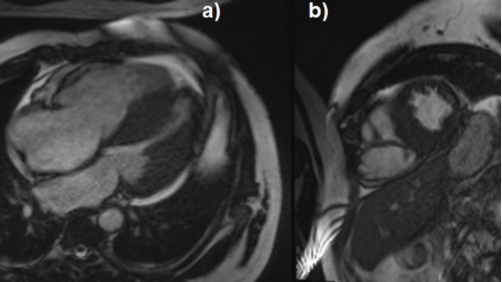
Figure 2. a) Cine CMR 4 chamber view showing asymmetric mid left ventricle wall thickening with reduced cavity size along with dilated right ventricle b) Short axis view confirming the above findings with 29 mm end diastolic wall thickening of septum and posterior inferior walls with dilated hypokinetic right ventricle.
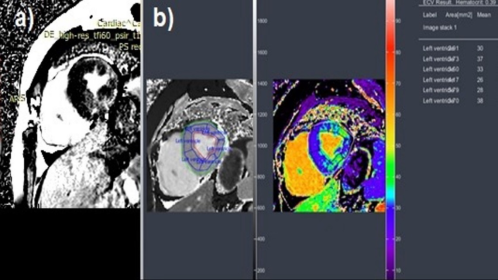
Figure 3. a) Post contrast late enhancement phase short axis view showing intramural areas of. Late enhancement in the inferior septal, posterior and anterior walls in left ventricle. b) ECV analysis image showing increased ECV of left ventricle.
Information

